马дёҠжіЁеҶҢпјҢз»“дәӨжӣҙеӨҡеҘҪеҸӢпјҢдә«з”ЁжӣҙеӨҡеҠҹиғҪпјҢи®©дҪ иҪ»жқҫзҺ©иҪ¬зӨҫеҢәгҖӮ
жӮЁйңҖиҰҒ зҷ»еҪ• жүҚеҸҜд»ҘдёӢиҪҪжҲ–жҹҘзңӢпјҢжІЎжңүиҙҰеҸ·пјҹз«ӢеҚіжіЁеҶҢ

x
жң¬её–жңҖеҗҺз”ұ иҘҝз“ңзұҪ дәҺ 2023-5-5 17:45 зј–иҫ‘ 0 e2 u) T/ O: n7 f- |' _
" Z9 L f! n3 {% f; \& Y3 {9 o) f, k3 z3 E- G+ b& {. z1 T
дҪңиҖ…пјҡж·јж·ј
# X) y1 G0 `; e. m* t# j; c# c. P- e! P$ a2 p8 ]# j2 s
иӣӢзҷҪиҙЁжҳҜз”ҹе‘Ҫзҡ„еҹәзЎҖпјҢе®ғ们еҸӮдёҺдәҶз»ҶиғһеҶ…еӨ–зҡ„еҗ„з§Қз”ҹзү©еӯҰиҝҮзЁӢпјҢеҰӮдҝЎеҸ·дј еҜјгҖҒд»Ји°ўгҖҒз»Ҷиғһе‘ЁжңҹгҖҒеҹәеӣ иЎЁиҫҫзӯүгҖӮ然иҖҢпјҢжңүдәӣиӣӢзҷҪиҙЁеңЁжӯЈеёёжғ…еҶөдёӢжҳҜжңүзӣҠзҡ„пјҢдҪҶеңЁз–ҫз—…зҠ¶жҖҒдёӢпјҢеҰӮзҷҢз—ҮпјҢе®ғ们дјҡеҸҳеҫ—жңүе®іпјҢдҝғиҝӣиӮҝзҳӨзҡ„з”ҹй•ҝе’Ңжү©ж•ЈгҖӮеӣ жӯӨпјҢжҠ‘еҲ¶жҲ–ж¶ҲйҷӨиҝҷдәӣжңүе®іиӣӢзҷҪиҙЁжҳҜдёҖз§Қжңүж•Ҳзҡ„жІ»з–—зӯ–з•ҘгҖӮ * g# n0 e7 y8 `# Q
дј з»ҹзҡ„иҚҜзү©ејҖеҸ‘ж–№жі•дё»иҰҒжҳҜеҜ»жүҫиғҪеӨҹдёҺзӣ®ж ҮиӣӢзҷҪиҙЁз»“еҗҲ并йҳ»ж–ӯе…¶еҠҹиғҪзҡ„е°ҸеҲҶеӯҗеҢ–еҗҲзү©пјҢиҝҷдәӣеҢ–еҗҲзү©иў«з§°дёәжҠ‘еҲ¶еүӮгҖӮдҪҶжҳҜпјҢжҠ‘еҲ¶еүӮеӯҳеңЁдёҖдәӣеұҖйҷҗжҖ§пјҢеҰӮйҖүжӢ©жҖ§дёҚй«ҳгҖҒеүҜдҪңз”ЁеӨҡгҖҒе®№жҳ“иҖҗиҚҜзӯүгҖӮдёәдәҶе…ӢжңҚиҝҷдәӣеұҖйҷҗжҖ§пјҢиҝ‘е№ҙжқҘеҮәзҺ°дәҶдёҖз§Қж–°еһӢзҡ„иҚҜзү©ејҖеҸ‘ж–№жі•пјҢеҚіеҲ©з”Ёз»ҶиғһеҶ…иҮӘиә«зҡ„иӣӢзҷҪиҙЁйҷҚи§Јзі»з»ҹжқҘжё…йҷӨзӣ®ж ҮиӣӢзҷҪиҙЁпјҢиҝҷдәӣиҚҜзү©иў«з§°дёәиӣӢзҷҪйҷҚи§ЈеүӮгҖӮ . @6 }/ r2 |( Q5 y
иӣӢзҷҪйҷҚи§ЈеүӮжҳҜдёҖзұ»еҸҢеҠҹиғҪжҲ–еӨҡеҠҹиғҪзҡ„е°ҸеҲҶеӯҗеҢ–еҗҲзү©пјҢе®ғ们иғҪеӨҹеҗҢж—¶дёҺзӣ®ж ҮиӣӢзҷҪиҙЁе’ҢдёҖдёӘжіӣзҙ иҝһжҺҘй…¶пјҲE3 ligaseпјүз»“еҗҲпјҢд»ҺиҖҢиҜұеҜјзӣ®ж ҮиӣӢзҷҪиҙЁиў«жіӣзҙ еҢ–并йҖҒе…ҘиӣӢзҷҪй…¶дҪ“иҝӣиЎҢйҷҚи§ЈгҖӮ
7 |: F% d, w, e. Jжіӣзҙ еҢ–жҳҜдёҖз§Қзҝ»иҜ‘еҗҺдҝ®йҘ°иҝҮзЁӢпјҢеҚіеңЁиӣӢзҷҪиҙЁдёҠиҝһжҺҘдёҖдёӘжҲ–еӨҡдёӘжіӣзҙ еҲҶеӯҗпјҢд»ҺиҖҢж”№еҸҳиӣӢзҷҪиҙЁзҡ„зЁіе®ҡжҖ§гҖҒдә’дҪңжҖ§жҲ–е®ҡдҪҚгҖӮиӣӢзҷҪй…¶дҪ“жҳҜдёҖз§ҚеӨҡдәҡеҹәзҡ„еӨ§еҲҶеӯҗеӨҚеҗҲзү©пјҢе®ғиғҪеӨҹиҜҶеҲ«е№¶йҷҚи§Јиў«еӨҡжіӣзҙ еҢ–зҡ„иӣӢзҷҪиҙЁгҖӮйҖҡиҝҮиҝҷз§Қж–№ејҸпјҢиӣӢзҷҪйҷҚи§ЈеүӮеҸҜд»Ҙе®һзҺ°еҜ№зӣ®ж ҮиӣӢзҷҪиҙЁзҡ„е®Ңе…Ёжё…йҷӨпјҢиҖҢдёҚд»…д»…жҳҜжҡӮж—¶жҖ§зҡ„жҠ‘еҲ¶гҖӮ ! b" f0 x: {; F0 V6 R& j$ b; F o4 s4 b
еңЁд»Ҡе№ҙзҡ„AACRе№ҙдјҡдёҠжңүдёҚе°‘ж–°зҡ„иӣӢзҷҪйҷҚи§ЈеүӮйқўдё–пјҢи®©жҲ‘们дёҖиө·жқҘзңӢдёҖзңӢйғҪжңүе“ӘдәӣгҖӮ
8 ]: j0 a: i0 j; h' D5 t8 p Q/ V5 QASP-3082пјҡдёҖз§ҚеҸЈжңҚзҡ„KRAS G12DйҷҚи§ЈеүӮпјҢеҸҜйҷҚи§ЈKRAS G12DиӣӢзҷҪпјҢз”ЁдәҺжІ»з–—KRAS G12Dй©ұеҠЁзҡ„йқһе°Ҹз»ҶиғһиӮәзҷҢпјҲNSCLCпјүе’Ңе…¶д»–е®һдҪ“зҳӨгҖӮ 3 {; Q1 b! |1 s3 k: x$ U0 ?5 T
0 J" L. p4 D) I/ H( {- W
KRASжҳҜдёҖз§Қе°ҸGTPй…¶пјҢе®ғеңЁRAS/RAF/MEK/ERKдҝЎеҸ·йҖҡи·Ҝдёӯиө·зқҖйҮҚиҰҒдҪңз”ЁгҖӮиҜҘдҝЎеҸ·йҖҡи·ҜеңЁз»Ҷиғһеўһж®–гҖҒеҲҶеҢ–гҖҒиҝҒ移е’ҢеҮӢдәЎзӯүиҝҮзЁӢдёӯеҸ‘жҢҘзқҖе…ій”®дҪңз”ЁгҖӮ 5 F3 P- i% P' X
然иҖҢпјҢиҜҘдҝЎеҸ·йҖҡи·ҜеңЁеӨҡз§ҚзҷҢз—Үдёӯиў«ејӮеёёжҝҖжҙ»пјҢеҜјиҮҙиӮҝзҳӨзҡ„еҸ‘з”ҹе’ҢеҸ‘еұ•гҖӮе…¶дёӯпјҢжңҖеёёи§Ғзҡ„ејӮеёёжҝҖжҙ»жңәеҲ¶жҳҜKRASеҹәеӣ зҡ„зӘҒеҸҳпјҢе°Өе…¶жҳҜG12DзӘҒеҸҳпјҢеҜјиҮҙKRASиҺ·еҫ—жҢҒз»ӯзҡ„жҝҖй…¶жҙ»жҖ§гҖӮзӣ®еүҚпјҢKRASжҠ‘еҲ¶еүӮе·Із»ҸжҲҗдёәйқһе°Ҹз»ҶиғһиӮәзҷҢпјҲNSCLCпјүе’Ңе…¶д»–KRASй©ұеҠЁзҡ„е®һдҪ“зҳӨзҡ„жңүж•ҲжІ»з–—жүӢж®өпјҢдҪҶд№ҹеӯҳеңЁдёҖдәӣй—®йўҳпјҢеҰӮйҖүжӢ©жҖ§дёҚй«ҳгҖҒеүҜдҪңз”ЁеӨҡгҖҒиҖҗиҚҜжҖ§й«ҳзӯүгҖӮеӣ жӯӨпјҢйҷҚи§ЈKRASжҳҜдёҖз§ҚжҸҗй«ҳйҖүжӢ©жҖ§е’Ңе®үе…ЁжҖ§зҡ„зӯ–з•ҘгҖӮ8 u& @4 R5 A. ?* |3 J. u" w
1 z# R& d* l3 @( S. E# p' ~ASP-3082жҳҜдёҖз§ҚеҸЈжңҚзҡ„KRAS G12DйҷҚи§ЈеүӮпјҢе®ғйҖҡиҝҮдёҺCereblonпјҲCRBNпјүE3 ligaseз»“еҗҲпјҢиҜұеҜјKRAS G12DиӣӢзҷҪзҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈгҖӮ
3 Q$ z$ O9 u' c4 T( r% D0 q0 fеңЁдҪ“еӨ–е®һйӘҢдёӯпјҢASP-3082иғҪеӨҹжңүж•ҲйҷҚи§ЈKRAS G12DиӣӢзҷҪпјҢ并жҠ‘еҲ¶еӨҡз§ҚKRAS G12Dй©ұеҠЁзҡ„NSCLCе’Ңе…¶д»–е®һдҪ“зҳӨз»Ҷиғһзі»зҡ„еўһж®–е’Ңеӯҳжҙ»гҖӮеңЁдҪ“еҶ…е®һйӘҢдёӯпјҢASP-3082иғҪеӨҹеңЁе°Ҹйј жЁЎеһӢдёӯжҳҫи‘—йҷҚдҪҺиӮҝзҳӨдёӯKRAS G12DиӣӢзҷҪзҡ„ж°ҙе№іпјҢ并жҠ‘еҲ¶иӮҝзҳӨзҡ„з”ҹй•ҝгҖӮ
0 X% v u) @' y5 dдёҺзҺ°жңүзҡ„KRASжҠ‘еҲ¶еүӮзӣёжҜ”пјҢASP-3082е…·жңүжӣҙй«ҳзҡ„йҖүжӢ©жҖ§е’Ңе®үе…ЁжҖ§гҖӮзӣ®еүҚпјҢASP-3082жӯЈеңЁиҝӣиЎҢдёҙеәҠеүҚејҖеҸ‘пјҢйў„и®Ўе°ҶдәҺ2024е№ҙејҖе§ӢдёҙеәҠиҜ•йӘҢгҖӮ
( [% S3 o2 q t! }$ `3 RARV-766пјҡдёҖз§ҚеҸЈжңҚзҡ„йӣ„жҝҖзҙ еҸ—дҪ“пјҲARпјүPROTACпјҢеҸҜйҷҚи§ЈйҮҺз”ҹеһӢе’ҢзӘҒеҸҳеһӢARпјҢз”ЁдәҺжІ»з–—еҺ»еҠҝжҠөжҠ—жҖ§еүҚеҲ—и…әзҷҢпјҲCRPCпјүгҖӮ
2 l" G" b% k( y# m. T
/ u' Z* A' j7 Y p2 j' v3 Y( }& k9 \йӣ„жҝҖзҙ еҸ—дҪ“пјҲARпјүжҳҜдёҖз§Қж ёеҸ—дҪ“пјҢе®ғеңЁеүҚеҲ—и…әзҷҢзҡ„еҸ‘з”ҹе’ҢеҸ‘еұ•дёӯиө·зқҖе…ій”®дҪңз”ЁгҖӮзӣ®еүҚпјҢжҠ—йӣ„жҝҖзҙ з–—жі•жҳҜеүҚеҲ—и…әзҷҢзҡ„дё»иҰҒжІ»з–—жүӢж®өпјҢдҪҶеӨ§еӨҡж•°жӮЈиҖ…жңҖз»ҲдјҡеҮәзҺ°еҺ»еҠҝжҠөжҠ—жҖ§еүҚеҲ—и…әзҷҢпјҲCRPCпјүпјҢеҚіеңЁйӣ„жҝҖзҙ ж°ҙе№іжһҒдҪҺзҡ„жғ…еҶөдёӢд»ҚиғҪз”ҹй•ҝзҡ„еүҚеҲ—и…әзҷҢгҖӮCRPCзҡ„дё»иҰҒжңәеҲ¶д№ӢдёҖжҳҜARзҡ„иҝҮеәҰиЎЁиҫҫжҲ–зӘҒеҸҳпјҢеҜјиҮҙеҜ№жҠ—йӣ„жҝҖзҙ з–—жі•зҡ„иҖҗиҚҜжҖ§гҖӮеӣ жӯӨпјҢйҷҚи§ЈARжҳҜдёҖз§Қжңүж•Ҳзҡ„жІ»з–—CRPCзҡ„зӯ–з•ҘгҖӮ 0 r9 U$ S! i) V9 M% G2 v8 i9 U N
ARV-766жҳҜдёҖз§ҚеҸЈжңҚзҡ„AR PROTACпјҢе®ғйҖҡиҝҮдёҺVHL E3 ligaseз»“еҗҲпјҢиҜұеҜјARзҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈгҖӮеңЁдҪ“еӨ–е®һйӘҢдёӯпјҢARV-766иғҪеӨҹжңүж•ҲйҷҚи§ЈйҮҺз”ҹеһӢе’ҢзӘҒеҸҳеһӢпјҲеҰӮF876LгҖҒW742LзӯүпјүARпјҢ并жҠ‘еҲ¶еӨҡз§ҚCRPCз»Ҷиғһзі»зҡ„еўһж®–гҖӮеңЁдҪ“еҶ…е®һйӘҢдёӯпјҢARV-766иғҪеӨҹеңЁе°Ҹйј жЁЎеһӢдёӯжҳҫи‘—йҷҚдҪҺиӮҝзҳӨдёӯARзҡ„ж°ҙе№іпјҢ并жҠ‘еҲ¶иӮҝзҳӨзҡ„з”ҹй•ҝгҖӮдёҺзҺ°жңүзҡ„жҠ—йӣ„жҝҖзҙ иҚҜзү©зӣёжҜ”пјҢARV-766е…·жңүжӣҙејәзҡ„иҚҜж•Ҳе’ҢжӣҙдҪҺзҡ„жҜ’жҖ§гҖӮзӣ®еүҚпјҢARV-766жӯЈеңЁиҝӣиЎҢдёҙеәҠеүҚејҖеҸ‘пјҢйў„и®Ўе°ҶдәҺ2024е№ҙејҖе§ӢдёҙеәҠиҜ•йӘҢгҖӮ
9 {* p: D' e2 E& C7 d( u; R" }( HNX-2127пјҡдёҖз§ҚеҸЈжңҚзҡ„еёғйІҒйЎҝй…Әж°Ёй…ёжҝҖй…¶пјҲBTKпјүйҷҚи§ЈеүӮпјҢеҸҜйҷҚи§ЈйҮҺз”ҹеһӢе’ҢC481SзӘҒеҸҳеһӢBTKпјҢз”ЁдәҺжІ»з–—еӨҚеҸ‘жҲ–йҡҫжІ»жҖ§Bз»ҶиғһжҒ¶жҖ§иӮҝзҳӨгҖӮ 5 L3 H: `& f' J# A' ~$ @2 {
7 A: o3 I- O* L% l2 I
еёғйІҒйЎҝй…Әж°Ёй…ёжҝҖй…¶пјҲBTKпјүжҳҜдёҖз§ҚйқһеҸ—дҪ“еһӢй…Әж°Ёй…ёжҝҖй…¶пјҢе®ғеңЁBз»ҶиғһеҸ—дҪ“пјҲBCRпјүдҝЎеҸ·йҖҡи·Ҝдёӯиө·зқҖйҮҚиҰҒдҪңз”ЁгҖӮBCRдҝЎеҸ·йҖҡи·ҜеңЁBз»ҶиғһжҒ¶жҖ§иӮҝзҳӨпјҢеҰӮж…ўжҖ§ж·Ӣе·ҙз»ҶиғһзҷҪиЎҖз—…пјҲCLLпјүгҖҒж»ӨжіЎжҖ§ж·Ӣе·ҙзҳӨпјҲFLпјүгҖҒиҫ№зјҳеҢәж·Ӣе·ҙзҳӨпјҲMZLпјүгҖҒе№јзЁҡеһӢж·Ӣе·ҙзҳӨпјҲMCLпјүзӯүдёӯеҸ‘жҢҘзқҖе…ій”®дҪңз”ЁгҖӮзӣ®еүҚпјҢBTKжҠ‘еҲ¶еүӮе·Із»ҸжҲҗдёәиҝҷдәӣиӮҝзҳӨзҡ„жңүж•ҲжІ»з–—жүӢж®өпјҢдҪҶд№ҹеӯҳеңЁдёҖдәӣй—®йўҳпјҢеҰӮдёҚиүҜеҸҚеә”гҖҒдёҚе®Ңе…ЁеҸҚеә”гҖҒеӨҚеҸ‘е’ҢиҖҗиҚҜжҖ§зӯүгҖӮ
4 ?$ [1 x" \4 w4 Z$ A( f8 ~& zе…¶дёӯпјҢжңҖеёёи§Ғзҡ„иҖҗиҚҜжңәеҲ¶жҳҜBTK C481SзӘҒеҸҳпјҢеҜјиҮҙBTKжҠ‘еҲ¶еүӮеӨұеҺ»з»“еҗҲиғҪеҠӣгҖӮеӣ жӯӨпјҢйҷҚи§ЈBTKжҳҜдёҖз§Қе…ӢжңҚиҖҗиҚҜжҖ§е’ҢжҸҗй«ҳжІ»з–—ж•Ҳжһңзҡ„зӯ–з•ҘгҖӮ ( ~) q; Y1 l- S/ ^8 |$ g( \0 p
NX-2127жҳҜдёҖз§ҚеҸЈжңҚзҡ„BTKйҷҚи§ЈеүӮпјҢе®ғйҖҡиҝҮдёҺCereblonпјҲCRBNпјүE3 ligaseз»“еҗҲпјҢиҜұеҜјBTKзҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈгҖӮеңЁдҪ“еӨ–е®һйӘҢдёӯпјҢNX-2127иғҪеӨҹжңүж•ҲйҷҚи§ЈйҮҺз”ҹеһӢе’ҢC481SзӘҒеҸҳеһӢBTKпјҢ并жҠ‘еҲ¶еӨҡз§ҚBз»ҶиғһжҒ¶жҖ§иӮҝзҳӨз»Ҷиғһзі»зҡ„еўһж®–е’Ңеӯҳжҙ»гҖӮеңЁдҪ“еҶ…е®һйӘҢдёӯпјҢNX-2127иғҪеӨҹеңЁе°Ҹйј жЁЎеһӢдёӯжҳҫи‘—йҷҚдҪҺиӮҝзҳӨдёӯBTKзҡ„ж°ҙе№іпјҢ并жҠ‘еҲ¶иӮҝзҳӨзҡ„з”ҹй•ҝгҖӮдёҺзҺ°жңүзҡ„BTKжҠ‘еҲ¶еүӮзӣёжҜ”пјҢNX-2127е…·жңүжӣҙејәзҡ„иҚҜж•Ҳе’ҢжӣҙдҪҺзҡ„жҜ’жҖ§гҖӮ
, h1 k' U- g; S! X7 g1 L1 fзӣ®еүҚпјҢNX-2127жӯЈеңЁиҝӣиЎҢдёҙеәҠеүҚејҖеҸ‘пјҢйў„и®Ўе°ҶдәҺ2024е№ҙејҖе§ӢдёҙеәҠиҜ•йӘҢгҖӮ 4 Q0 H3 n( n* Q1 u9 `7 L7 l! Y
CFT1946пјҡдёҖз§ҚеҸЈжңҚзҡ„BRAF PROTACпјҢеҸҜйҷҚи§ЈйҮҺз”ҹеһӢе’ҢV600EзӘҒеҸҳеһӢBRAFпјҢз”ЁдәҺжІ»з–—й»‘иүІзҙ зҳӨе’Ңе…¶д»–BRAFй©ұеҠЁзҡ„е®һдҪ“зҳӨгҖӮ
1 h0 ]) f; g8 X( ]& C3 @+ e: @+ N6 a/ h/ j. h9 }6 j
BRAFжҳҜдёҖз§Қдёқж°Ёй…ё/иӢҸж°Ёй…ёжҝҖй…¶пјҢе®ғеңЁRAS/RAF/MEK/ERKдҝЎеҸ·йҖҡи·Ҝдёӯиө·зқҖйҮҚиҰҒдҪңз”ЁгҖӮиҜҘдҝЎеҸ·йҖҡи·ҜеңЁз»Ҷиғһеўһж®–гҖҒеҲҶеҢ–гҖҒиҝҒ移е’ҢеҮӢдәЎзӯүиҝҮзЁӢдёӯеҸ‘жҢҘзқҖе…ій”®дҪңз”ЁгҖӮ然иҖҢпјҢиҜҘдҝЎеҸ·йҖҡи·ҜеңЁеӨҡз§ҚзҷҢз—Үдёӯиў«ејӮеёёжҝҖжҙ»пјҢеҜјиҮҙиӮҝзҳӨзҡ„еҸ‘з”ҹе’ҢеҸ‘еұ•гҖӮе…¶дёӯпјҢжңҖеёёи§Ғзҡ„ејӮеёёжҝҖжҙ»жңәеҲ¶жҳҜBRAFеҹәеӣ зҡ„зӘҒеҸҳпјҢе°Өе…¶жҳҜV600EзӘҒеҸҳпјҢеҜјиҮҙBRAFиҺ·еҫ—жҢҒз»ӯзҡ„жҝҖй…¶жҙ»жҖ§гҖӮ
- ]3 ?; s( l% a( n5 J& Hзӣ®еүҚпјҢBRAFжҠ‘еҲ¶еүӮе·Із»ҸжҲҗдёәй»‘иүІзҙ зҳӨе’Ңе…¶д»–BRAFй©ұеҠЁзҡ„е®һдҪ“зҳӨзҡ„жңүж•ҲжІ»з–—жүӢж®өпјҢдҪҶд№ҹеӯҳеңЁдёҖдәӣй—®йўҳпјҢеҰӮдёҚиүҜеҸҚеә”гҖҒдёҚе®Ңе…ЁеҸҚеә”гҖҒеӨҚеҸ‘е’ҢиҖҗиҚҜжҖ§зӯүгҖӮе…¶дёӯпјҢжңҖеёёи§Ғзҡ„иҖҗиҚҜжңәеҲ¶жҳҜMEKжҲ–ERKдёҠжёёжҲ–дёӢжёёдҝЎеҸ·йҖҡи·Ҝзҡ„еҶҚжҝҖжҙ»пјҢеҜјиҮҙеҜ№BRAFжҠ‘еҲ¶еүӮзҡ„йҖғйҖёгҖӮеӣ жӯӨпјҢйҷҚи§ЈBRAFжҳҜдёҖз§Қе…ӢжңҚиҖҗиҚҜжҖ§е’ҢжҸҗй«ҳжІ»з–—ж•Ҳжһңзҡ„зӯ–з•ҘгҖӮ ( F t& Z8 }8 T! S6 ^
CFT1946жҳҜдёҖз§ҚеҸЈжңҚзҡ„BRAF PROTACпјҢе®ғйҖҡиҝҮдёҺVHL E3 ligaseз»“еҗҲпјҢиҜұеҜјBRAFзҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈгҖӮеңЁдҪ“еӨ–е®һйӘҢдёӯпјҢCFT1946иғҪеӨҹжңүж•ҲйҷҚи§ЈйҮҺз”ҹеһӢе’ҢV600EзӘҒеҸҳеһӢBRAFпјҢ并жҠ‘еҲ¶еӨҡз§Қй»‘иүІзҙ зҳӨе’Ңе…¶д»–BRAFй©ұеҠЁзҡ„е®һдҪ“зҳӨз»Ҷиғһзі»зҡ„еўһж®–е’Ңеӯҳжҙ»гҖӮ
( x- x% t @: f" |/ V3 kеңЁдҪ“еҶ…е®һйӘҢдёӯпјҢCFT1946иғҪеӨҹеңЁе°Ҹйј жЁЎеһӢдёӯжҳҫи‘—йҷҚдҪҺиӮҝзҳӨдёӯBRAFзҡ„ж°ҙе№іпјҢ并жҠ‘еҲ¶иӮҝзҳӨзҡ„з”ҹй•ҝгҖӮдёҺзҺ°жңүзҡ„BRAFжҠ‘еҲ¶еүӮзӣёжҜ”пјҢCFT1946е…·жңүжӣҙејәзҡ„иҚҜж•Ҳе’ҢжӣҙдҪҺзҡ„жҜ’жҖ§гҖӮзӣ®еүҚпјҢCFT1946жӯЈеңЁиҝӣиЎҢдёҙеәҠеүҚејҖеҸ‘пјҢйў„и®Ўе°ҶдәҺ2024е№ҙејҖе§ӢдёҙеәҠиҜ•йӘҢгҖӮ
: f: D- c3 d8 z! k, S' c' BH001пјҡдёҖз§ҚеҸЈжңҚзҡ„RIPTACиҚҜзү©пјҢеҸҜиҜұеҜјеүҚеҲ—и…әзҷҢзү№ејӮжҖ§иӣӢзҷҪйӣ„жҝҖзҙ еҸ—дҪ“пјҲARпјүе’Ңеҝ…йңҖиҪ¬еҪ•и°ғиҠӮеӣ еӯҗд№Ӣй—ҙзҡ„дёүиҒҡдҪ“еӨҚеҗҲзү©еҪўжҲҗпјҢд»ҺиҖҢеҜјиҮҙеүҚеҲ—и…әзҷҢз»ҶиғһйҖүжӢ©жҖ§жӯ»дәЎгҖӮ & A: X. W4 V4 o9 N9 S
7 P2 V$ _, `% U" l( I5 D! cRIPTACsпјҲReceptor Interacting Protein TACsпјүжҳҜдёҖзұ»ж–°еһӢзҡ„иӣӢзҷҪйҷҚи§ЈеүӮпјҢе®ғ们иғҪеӨҹиҜұеҜјдёӨз§ҚдёҚеҗҢзұ»еһӢзҡ„иӣӢзҷҪиҙЁд№Ӣй—ҙеҪўжҲҗеӨҚеҗҲзү©пјҢ并е°Ҷе…¶йҖҒе…ҘиӣӢзҷҪй…¶дҪ“иҝӣиЎҢйҷҚи§ЈгҖӮдёҺPROTACsдёҚеҗҢпјҢRIPTACsдёҚйңҖиҰҒдёҺE3 ligaseз»“еҗҲпјҢиҖҢжҳҜеҲ©з”ЁдёҖз§ҚеҸ—дҪ“зӣёдә’дҪңз”ЁиӣӢзҷҪпјҲRIPпјүжқҘд»ӢеҜјеӨҚеҗҲзү©еҪўжҲҗгҖӮRIPжҳҜдёҖз§ҚиғҪеӨҹдёҺзү№е®ҡеҸ—дҪ“з»“еҗҲ并и°ғиҠӮе…¶еҠҹиғҪзҡ„иӣӢзҷҪиҙЁпјҢеҰӮйӣ„жҝҖзҙ еҸ—дҪ“пјҲARпјүгҖҒйӣҢжҝҖзҙ еҸ—дҪ“пјҲERпјүгҖҒеӯ•жҝҖзҙ еҸ—дҪ“пјҲPRпјүзӯүгҖӮйҖҡиҝҮе°ҶRIPдёҺеҸҰдёҖз§Қзӣ®ж ҮиӣӢзҷҪиҙЁиҝһжҺҘиө·жқҘпјҢRIPTACsеҸҜд»ҘдҪҝиҝҷдёӨз§ҚиӣӢзҷҪиҙЁеҪўжҲҗдёҖдёӘдёҚзЁіе®ҡзҡ„дёүиҒҡдҪ“еӨҚеҗҲзү©пјҢ并被иӣӢзҷҪй…¶дҪ“иҜҶеҲ«е’ҢйҷҚи§ЈгҖӮ
% R) z6 i, [4 z0 qH001жҳҜдёҖз§ҚеҸЈжңҚзҡ„RIPTACиҚҜзү©пјҢе®ғйҖҡиҝҮе°ҶARдёҺдёҖдёӘеҝ…йңҖзҡ„иҪ¬еҪ•и°ғиҠӮеӣ еӯҗпјҲTRFпјүиҝһжҺҘиө·жқҘпјҢиҜұеҜјеүҚеҲ—и…әзҷҢзү№ејӮжҖ§иӣӢзҷҪARе’ҢTRFд№Ӣй—ҙзҡ„дёүиҒҡдҪ“еӨҚеҗҲзү©еҪўжҲҗпјҢ并е°Ҷе…¶йҖҒе…ҘиӣӢзҷҪй…¶дҪ“иҝӣиЎҢйҷҚи§ЈгҖӮ 4 \+ s+ n' D3 s3 w- B) f
еңЁдҪ“еӨ–е®һйӘҢдёӯпјҢH001иғҪеӨҹжңүж•ҲйҷҚи§ЈARе’ҢTRFпјҢ并жҠ‘еҲ¶еӨҡз§ҚеүҚеҲ—и…әзҷҢз»Ҷиғһзі»зҡ„еўһж®–е’Ңеӯҳжҙ»гҖӮеңЁдҪ“еҶ…е®һйӘҢдёӯпјҢH001иғҪеӨҹеңЁе°Ҹйј жЁЎеһӢдёӯжҳҫи‘—йҷҚдҪҺиӮҝзҳӨдёӯARе’ҢTRFзҡ„ж°ҙе№іпјҢ并жҠ‘еҲ¶иӮҝзҳӨзҡ„з”ҹй•ҝгҖӮ
0 c. N* L3 B0 w7 j& D: \дёҺзҺ°жңүзҡ„ARжҠ‘еҲ¶еүӮжҲ–йҷҚи§ЈеүӮзӣёжҜ”пјҢH001е…·жңүжӣҙй«ҳзҡ„йҖүжӢ©жҖ§е’Ңе®үе…ЁжҖ§пјҢеӣ дёәе®ғеҸӘеңЁеүҚеҲ—и…әзҷҢз»Ҷиғһдёӯиө·дҪңз”ЁпјҢиҖҢдёҚеҪұе“ҚжӯЈеёёз»„з»Үдёӯзҡ„ARе’ҢTRFгҖӮзӣ®еүҚпјҢH001жӯЈеңЁиҝӣиЎҢдёҙеәҠеүҚејҖеҸ‘пјҢйў„и®Ўе°ҶдәҺ2024е№ҙејҖе§ӢдёҙеәҠиҜ•йӘҢгҖӮ ' M! J+ `& W) p; z3 S) y5 T+ G G
MRT-2359пјҡдёҖз§ҚеҸЈжңҚзҡ„GSPT1еҲҶеӯҗиғ¶йҷҚи§ЈеүӮпјҲMGDпјүпјҢеҸҜйҖҡиҝҮCRBNд»ӢеҜјGSPT1зҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈпјҢз”ЁдәҺжІ»з–—MYCй©ұеҠЁзҡ„е®һдҪ“зҳӨгҖӮ I+ a$ i% T9 W* ~2 x0 }1 g# W
' V3 O' y, a) u% ~. TGSPT1жҳҜдёҖз§ҚиҪ¬еҪ•еҗҺдҝ®йҘ°еӣ еӯҗпјҢе®ғиғҪеӨҹдёҺMYCиӣӢзҷҪз»“еҗҲ并稳е®ҡе…¶з»“жһ„пјҢд»ҺиҖҢеўһејәе…¶иҪ¬еҪ•жҙ»жҖ§гҖӮMYCжҳҜдёҖз§ҚиҪ¬еҪ•еӣ еӯҗпјҢе®ғиғҪеӨҹи°ғжҺ§еӨҡз§Қеҹәеӣ зҡ„иЎЁиҫҫпјҢеҰӮз»Ҷиғһе‘ЁжңҹгҖҒд»Ји°ўгҖҒз»Ҷиғһжӯ»дәЎзӯүгҖӮ 2 w; \0 P/ J2 h; {9 u* V
然иҖҢпјҢеңЁеӨҡз§ҚзҷҢз—ҮдёӯпјҢMYCиў«иҝҮеәҰиЎЁиҫҫжҲ–жү©еўһпјҢеҜјиҮҙиӮҝзҳӨзҡ„еҸ‘з”ҹе’ҢеҸ‘еұ•гҖӮзӣ®еүҚпјҢMYCжҠ‘еҲ¶еүӮе·Із»ҸжҲҗдёәMYCй©ұеҠЁзҡ„е®һдҪ“зҳӨзҡ„жңүж•ҲжІ»з–—жүӢж®өпјҢдҪҶд№ҹеӯҳеңЁдёҖдәӣй—®йўҳпјҢеҰӮйҖүжӢ©жҖ§дёҚй«ҳгҖҒеүҜдҪңз”ЁеӨҡгҖҒе®№жҳ“иҖҗиҚҜзӯүгҖӮеӣ жӯӨпјҢйҷҚи§ЈGSPT1жҳҜдёҖз§ҚжҠ‘еҲ¶MYCдҝЎеҸ·йҖҡи·Ҝзҡ„зӯ–з•ҘгҖӮ 1 E1 t+ E+ e$ C
MRT-2359жҳҜдёҖз§ҚеҸЈжңҚзҡ„GSPT1еҲҶеӯҗиғ¶йҷҚи§ЈеүӮпјҲMGDпјүпјҢе®ғйҖҡиҝҮдёҺCereblonпјҲCRBNпјүE3 ligaseз»“еҗҲпјҢиҜұеҜјGSPT1зҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈгҖӮеңЁдҪ“еӨ–е®һйӘҢдёӯпјҢMRT-2359иғҪеӨҹжңүж•ҲйҷҚи§ЈGSPT1пјҢ并жҠ‘еҲ¶еӨҡз§ҚMYCй©ұеҠЁзҡ„е®һдҪ“зҳӨз»Ҷиғһзі»зҡ„еўһж®–е’Ңеӯҳжҙ»гҖӮ
7 `9 }8 M0 t# o5 wеңЁдҪ“еҶ…е®һйӘҢдёӯпјҢMRT-2359иғҪеӨҹеңЁе°Ҹйј жЁЎеһӢдёӯжҳҫи‘—йҷҚдҪҺиӮҝзҳӨдёӯGSPT1зҡ„ж°ҙе№іпјҢ并жҠ‘еҲ¶иӮҝзҳӨзҡ„з”ҹй•ҝгҖӮдёҺзҺ°жңүзҡ„MYCжҠ‘еҲ¶еүӮзӣёжҜ”пјҢMRT-2359е…·жңүжӣҙй«ҳзҡ„йҖүжӢ©жҖ§е’Ңе®үе…ЁжҖ§пјҢеӣ дёәе®ғеҸӘй’ҲеҜ№GSPT1пјҢиҖҢдёҚеҪұе“Қе…¶д»–MYCз»“еҗҲиӣӢзҷҪжҲ–иҪ¬еҪ•еӣ еӯҗгҖӮзӣ®еүҚпјҢMRT-2359жӯЈеңЁиҝӣиЎҢдёҙеәҠеүҚејҖеҸ‘пјҢйў„и®Ўе°ҶдәҺ2024е№ҙејҖе§ӢдёҙеәҠиҜ•йӘҢгҖӮ
2 o) d8 B/ J) |2 ]# f; p9 P4 QSCR-9140пјҡдёҖз§ҚеҸЈжңҚзҡ„SMARCA2иӣӢзҷҪйҷҚи§ЈеүӮпјҢеҸҜйҖҡиҝҮCRBNд»ӢеҜјSMARCA2зҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈпјҢз”ЁдәҺжІ»з–—SMARCA4зјәеӨұзҡ„вҖңеҗҲжҲҗиҮҙжӯ»вҖқиӮҝзҳӨгҖӮ ! G( }% N' H: o6 h3 y7 L* S
: N+ c( e( {5 c' ?
SMARCA2е’ҢSMARCA4жҳҜдёӨз§Қжҹ“иүІиҙЁйҮҚеЎ‘еӣ еӯҗпјҢе®ғ们еұһдәҺSWI/SNFеӨҚеҗҲзү©зҡ„ж ёеҝғжҲҗе‘ҳпјҢиғҪеӨҹи°ғиҠӮжҹ“иүІиҙЁз»“жһ„е’Ңеҹәеӣ иЎЁиҫҫгҖӮSWI/SNFеӨҚеҗҲзү©еңЁз»ҶиғһеҲҶеҢ–гҖҒеўһж®–гҖҒиҝҒ移е’ҢеҮӢдәЎзӯүиҝҮзЁӢдёӯеҸ‘жҢҘзқҖйҮҚиҰҒдҪңз”ЁгҖӮ
( K. X0 R: o3 ^8 e; o然иҖҢпјҢеңЁеӨҡз§ҚзҷҢз—ҮдёӯпјҢSWI/SNFеӨҚеҗҲзү©иў«зӘҒеҸҳжҲ–зјәеӨұпјҢеҜјиҮҙжҹ“иүІиҙЁйҮҚеЎ‘еҠҹиғҪдё§еӨұе’ҢиӮҝзҳӨзҡ„еҸ‘з”ҹе’ҢеҸ‘еұ•гҖӮе…¶дёӯпјҢжңҖеёёи§Ғзҡ„зӘҒеҸҳжҲ–зјәеӨұжңәеҲ¶жҳҜSMARCA4еҹәеӣ зҡ„зјәеӨұжҲ–жІүй»ҳпјҢеҜјиҮҙSMARCA4иӣӢзҷҪзјәд№ҸгҖӮ
/ m. y& R4 b- X8 O+ ]7 c0 T4 Nзӣ®еүҚпјҢй’ҲеҜ№SMARCA4зјәеӨұзҡ„иӮҝзҳӨжІЎжңүзү№ејӮжҖ§зҡ„жІ»з–—жүӢж®өпјҢдҪҶжңүдёҖз§ҚеҸҜиғҪзҡ„зӯ–з•ҘжҳҜеҲ©з”ЁвҖңеҗҲжҲҗиҮҙжӯ»вҖқзҡ„еҺҹзҗҶпјҢеҚіеҗҢж—¶йқ¶еҗ‘еҸҰдёҖз§ҚдёҺSMARCA4еҠҹиғҪйҮҚеҸ зҡ„иӣӢзҷҪиҙЁпјҢеҰӮSMARCA2пјҢд»ҺиҖҢеҜјиҮҙиӮҝзҳӨз»Ҷиғһжӯ»дәЎгҖӮ
9 a0 T0 n' L: \- V! {1 sSCR-9140жҳҜдёҖз§ҚеҸЈжңҚзҡ„SMARCA2иӣӢзҷҪйҷҚи§ЈеүӮпјҢе®ғйҖҡиҝҮдёҺCereblonпјҲCRBNпјүE3 ligaseз»“еҗҲпјҢиҜұеҜјSMARCA2зҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈгҖӮеңЁдҪ“еӨ–е®һйӘҢдёӯпјҢSCR-9140иғҪеӨҹжңүж•ҲйҷҚи§ЈSMARCA2пјҢ并жҠ‘еҲ¶еӨҡз§ҚSMARCA4зјәеӨұзҡ„вҖңеҗҲжҲҗиҮҙжӯ»вҖқиӮҝзҳӨз»Ҷиғһзі»зҡ„еўһж®–е’Ңеӯҳжҙ»гҖӮ
0 a, g. D! G8 m/ P5 ~( yеңЁдҪ“еҶ…е®һйӘҢдёӯпјҢSCR-9140иғҪеӨҹеңЁе°Ҹйј жЁЎеһӢдёӯжҳҫи‘—йҷҚдҪҺиӮҝзҳӨдёӯSMARCA2зҡ„ж°ҙе№іпјҢ并жҠ‘еҲ¶иӮҝзҳӨзҡ„з”ҹй•ҝгҖӮдёҺзҺ°жңүзҡ„SWI/SNFеӨҚеҗҲзү©жҠ‘еҲ¶еүӮзӣёжҜ”пјҢSCR-9140е…·жңүжӣҙй«ҳзҡ„йҖүжӢ©жҖ§е’Ңе®үе…ЁжҖ§пјҢеӣ дёәе®ғеҸӘй’ҲеҜ№SMARCA2пјҢиҖҢдёҚеҪұе“Қе…¶д»–SWI/SNFеӨҚеҗҲзү©жҲҗе‘ҳжҲ–еҠҹиғҪгҖӮзӣ®еүҚпјҢSCR-9140жӯЈеңЁиҝӣиЎҢдёҙеәҠеүҚејҖеҸ‘пјҢйў„и®Ўе°ҶдәҺ2024е№ҙејҖе§ӢдёҙеәҠиҜ•йӘҢгҖӮ
3 D: y: Q9 B ^3 f* nжҖ»д№ӢпјҢиӣӢзҷҪйҷҚи§ЈеүӮжҳҜдёҖз§Қе…·жңүеҲӣж–°жҖ§е’ҢеүҚжҷҜжҖ§зҡ„иҚҜзү©ејҖеҸ‘ж–№жі•пјҢе®ғ们иғҪеӨҹйҖҡиҝҮиҜұеҜјзӣ®ж ҮиӣӢзҷҪиҙЁзҡ„жіӣзҙ еҢ–е’ҢйҷҚи§ЈжқҘе®һзҺ°еҜ№зҷҢз—Үзҡ„жІ»з–—гҖӮ $ J. R* m( W+ @
иӣӢзҷҪйҷҚи§ЈеүӮзӣёжҜ”дј з»ҹзҡ„жҠ‘еҲ¶еүӮпјҢе…·жңүжӣҙеӨҡзҡ„дјҳеҠҝе’ҢжҪңеҠӣпјҢеҰӮиғҪеӨҹж”»е…ӢдёҖдәӣйҡҫд»ҘжҠ‘еҲ¶зҡ„зӣ®ж ҮгҖҒиғҪеӨҹе®һзҺ°еҜ№зӣ®ж ҮиӣӢзҷҪиҙЁзҡ„жҢҒз»ӯе’ҢеҪ»еә•жё…йҷӨгҖҒиғҪеӨҹеҲ©з”ЁдёҚеҗҢзұ»еһӢзҡ„E3 ligaseжқҘи°ғиҠӮдёҚеҗҢз»„з»ҮжҲ–з»Ҷиғһдёӯзҡ„зӣ®ж ҮиӣӢзҷҪиҙЁж°ҙе№ігҖҒиғҪеӨҹйҖҡиҝҮеҪұе“Қзӣ®ж ҮиӣӢзҷҪиҙЁжүҖеңЁзҡ„дҝЎеҸ·йҖҡи·ҜжҲ–зҪ‘з»ңжқҘдә§з”ҹеҚҸеҗҢжҲ–жӢ®жҠ—ж•Ҳеә”зӯүгҖӮ
: F; ~& c0 d1 T: y& a0 o然иҖҢпјҢиӣӢзҷҪйҷҚи§ЈеүӮд№ҹеӯҳеңЁдёҖдәӣжҢ‘жҲҳе’Ңй—®йўҳпјҢеҰӮйңҖиҰҒжҸҗй«ҳдёҺE3 ligaseе’Ңзӣ®ж ҮиӣӢзҷҪиҙЁзҡ„дәІе’ҢеҠӣе’Ңзү№ејӮжҖ§гҖҒйңҖиҰҒдјҳеҢ–иӣӢзҷҪйҷҚи§ЈеүӮзҡ„з»“жһ„е’ҢжҖ§иҙЁгҖҒйңҖиҰҒиҜ„дј°иӣӢзҷҪйҷҚи§ЈеүӮзҡ„иҚҜзҗҶеӯҰе’ҢжҜ’зҗҶеӯҰгҖҒйңҖиҰҒи®ҫи®ЎеҗҲзҗҶзҡ„дёҙеәҠиҜ•йӘҢж–№жЎҲзӯүгҖӮ 5 u' T% C5 n% M; n' o; _, V- t
еңЁд»Ҡе№ҙзҡ„AACRеӨ§дјҡдёҠеұ•зӨәзҡ„еҮ з§Қд»ЈиЎЁжҖ§зҡ„иӣӢзҷҪйҷҚи§ЈеүӮпјҢдёҚд»…еұ•зӨәдәҶиӣӢзҷҪйҷҚи§ЈеүӮеңЁзҷҢз—ҮжІ»з–—дёӯзҡ„еӨҡж ·жҖ§е’ҢеҸҜиғҪжҖ§пјҢд№ҹеҸҚжҳ дәҶиӣӢзҷҪйҷҚи§ЈеүӮеңЁзҷҢз—ҮжІ»з–—дёӯзҡ„дјҳеҠҝе’ҢжҢ‘жҲҳпјҢд»ҘеҸҠжңӘжқҘзҡ„еҸ‘еұ•ж–№еҗ‘гҖӮ ; W( t% B4 ?2 r" l
+ |1 L3 a* K( z7 u' Y) GеҸӮиҖғж–ҮзҢ® 2023 AACR * t P# h$ z0 l0 S
зӣёе…іеӣһйЎҫдёЁAACRзӣёе…іж–Үз« ) g( N4 O/ G( L* e& B# S3 m" f4 X
[color=var(--weui-LINK)]AACRйҖҹйҖ’дёЁе…Қз–«жІ»з–—вҖңеӨ№еҝғйҘјвҖқпјҢжҳҫи‘—йҷҚдҪҺеӨҚеҸ‘жҲ–жӯ»дәЎйЈҺйҷ© [color=var(--weui-LINK)]AACRйҖҹйҖ’дёЁеҘҘзҫҺжӢүе”‘еҪұе“ҚSotorasibеҗёж”¶пјҹдёҖжқҜвҖңеҝ«д№җж°ҙвҖқи§ЈеҶі [color=var(--weui-LINK)]AACRйҖҹйҖ’дёЁHER2йқ¶еҗ‘жІ»з–—пјҢADCгҖҒTKIж–°иҚҜйҪҗеҸ‘еұ•
. g" R/ O4 k+ G3 Q5 s | 



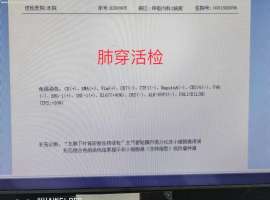
 гҖҗжұӮеҠ©гҖ‘иӮәи…әзҷҢв…ўA T1cN2M0пјҢиӮәеҶ…еҸҠ
2019е№ҙ11жңҲжүӢжңҜеҲҮйҷӨ
з—…зҗҶжҸҗзӨә:еҸіиӮәдёҠеҸ¶еҚ•еҸ‘иӮҝзҳӨпјҢеӨ§е°Ҹ2.5X1.5x1.5cmжөёж¶ҰжҖ§и…әзҷҢпјҢи…әжіЎ
гҖҗжұӮеҠ©гҖ‘иӮәи…әзҷҢв…ўA T1cN2M0пјҢиӮәеҶ…еҸҠ
2019е№ҙ11жңҲжүӢжңҜеҲҮйҷӨ
з—…зҗҶжҸҗзӨә:еҸіиӮәдёҠеҸ¶еҚ•еҸ‘иӮҝзҳӨпјҢеӨ§е°Ҹ2.5X1.5x1.5cmжөёж¶ҰжҖ§и…әзҷҢпјҢи…әжіЎ
 жұӮеҠ© KиҚҜе’Ңжӣҝйӣ·еҲ©зҸ еҰӮдҪ•йҖүжӢ©
зҲ¶дәІ9жңҲеҲҡжҹҘеҮәйқһе°Ҹз»ҶиғһдҪҺеҲҶеҢ–зҷҢдјҙйҡҸе·ҰиӮҫдёҠи…әе’Ң第10иғёжӨҺиҪ¬з§»PDL1й«ҳиЎЁиҫҫ(TPS=90%) SMARC
жұӮеҠ© KиҚҜе’Ңжӣҝйӣ·еҲ©зҸ еҰӮдҪ•йҖүжӢ©
зҲ¶дәІ9жңҲеҲҡжҹҘеҮәйқһе°Ҹз»ҶиғһдҪҺеҲҶеҢ–зҷҢдјҙйҡҸе·ҰиӮҫдёҠи…әе’Ң第10иғёжӨҺиҪ¬з§»PDL1й«ҳиЎЁиҫҫ(TPS=90%) SMARC
 жұӮеҠ©иҙҙпјҡж–°жүӢдёҖжһҡпјҢдёәдәҶзҲёзҲёеҸ‘еҮәжұӮеҠ©
зҲёзҲё2023е№ҙ11.28ж—ҘзЎ®иҜҠпјҢз—…зҗҶз»“жһңпјҡдјҙжңүиӮүзҳӨж ·зү№еҫҒзҡ„иӮәи…әзҷҢгҖӮжӮЈиҖ…жғ…еҶөпјҡз”·пјҢ52еІҒпјҢ12
жұӮеҠ©иҙҙпјҡж–°жүӢдёҖжһҡпјҢдёәдәҶзҲёзҲёеҸ‘еҮәжұӮеҠ©
зҲёзҲё2023е№ҙ11.28ж—ҘзЎ®иҜҠпјҢз—…зҗҶз»“жһңпјҡдјҙжңүиӮүзҳӨж ·зү№еҫҒзҡ„иӮәи…әзҷҢгҖӮжӮЈиҖ…жғ…еҶөпјҡз”·пјҢ52еІҒпјҢ12
 жҷҡжңҹиӮәзҷҢ13е№ҙйқ¶еҗ‘иҪ®жҚўд№Ӣи·ҜпјҢжҲ‘жҖ»з»“9
и®Іиҝ°иҖ…пјҡдёҚжҖ•иҫЈжӨ’дёҚжҖ•зҷҢж•ҙзҗҶиҖ…пјҡйӣӘжј“
дёҠзҜҮж–Үз« дёӯеҲҶдә«дәҶжҲ‘家13е№ҙжҠ—зҷҢз»ҸеҺҶд»ҘеҸҠеҝғжҖҒжҲҗй•ҝ
жҷҡжңҹиӮәзҷҢ13е№ҙйқ¶еҗ‘иҪ®жҚўд№Ӣи·ҜпјҢжҲ‘жҖ»з»“9
и®Іиҝ°иҖ…пјҡдёҚжҖ•иҫЈжӨ’дёҚжҖ•зҷҢж•ҙзҗҶиҖ…пјҡйӣӘжј“
дёҠзҜҮж–Үз« дёӯеҲҶдә«дәҶжҲ‘家13е№ҙжҠ—зҷҢз»ҸеҺҶд»ҘеҸҠеҝғжҖҒжҲҗй•ҝ
 еҲқиҜҠиғғзҷҢеҝ…зңӢпјҒжӮЈиҖ…家еұһжҖ»з»“29жқЎзңҹе®һ
дҪңиҖ…пјҡи“қе…Ҳз”ҹ
д»ҘдёӢд»…дёәдёҖдёӘжӮЈиҖ…家еұһзҡ„з»ҸйӘҢж•ҷи®ӯйӣҶй”ҰпјҢдёҖ家д№ӢиЁҖпјҢиӢҘжңүй”ҷи®№д№ӢеӨ„пјҢ敬иҜ·и°…
еҲқиҜҠиғғзҷҢеҝ…зңӢпјҒжӮЈиҖ…家еұһжҖ»з»“29жқЎзңҹе®һ
дҪңиҖ…пјҡи“қе…Ҳз”ҹ
д»ҘдёӢд»…дёәдёҖдёӘжӮЈиҖ…家еұһзҡ„з»ҸйӘҢж•ҷи®ӯйӣҶй”ҰпјҢдёҖ家д№ӢиЁҖпјҢиӢҘжңүй”ҷи®№д№ӢеӨ„пјҢ敬иҜ·и°…











 жҸҗеҚҮеҚЎ
жҸҗеҚҮеҚЎ зҪ®йЎ¶еҚЎ
зҪ®йЎ¶еҚЎ жІүй»ҳеҚЎ
жІүй»ҳеҚЎ е–§еҡЈеҚЎ
е–§еҡЈеҚЎ еҸҳиүІеҚЎ
еҸҳиүІеҚЎ еҚғж–ӨйЎ¶
еҚғж–ӨйЎ¶ жҳҫиә«еҚЎ
жҳҫиә«еҚЎ